Until recently, repetitive regions of the genome had been dismissed as junk DNA. However, it is now widely accepted that non-coding tandem repeats (TRs) hold highly valuable information that is important for regulating gene expression. To date, TRs have been implicated in causing over 30 rare diseases. Since large, novel TRs have been very difficult to identify in patients, we and others speculate that there may be many more rare diseases caused by repeat expansions. In this paper we describe the variation in repetitive DNA among 3,619 control individuals. We propose to use this as a comparative dataset to identify novel pathogenic repeats in disease genomes. This will be my mission for the remainder my PhD.
I joined Dr. Stephan Zuchner’s laboratory two years ago. He described my project as “high risk, high reward” which created mixed feelings of excitement and nervousness. Two years later, after long hours of developing my repeat expansion detection pipeline using the dataset described here, I am confident that I will find that needle in the haystack.
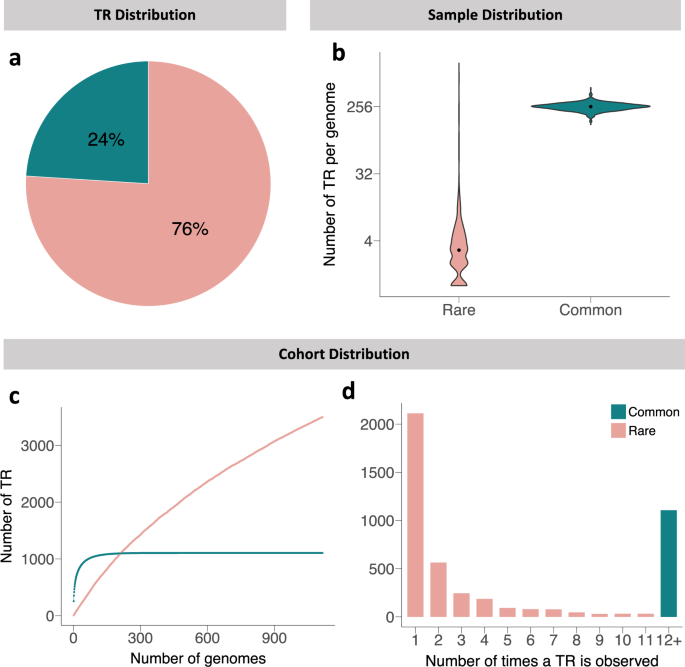
There is widespread interest among the scientific community in decoding repetitive DNA. However, this comes with many challenges. First and foremost is the obstacle of aligning repeat-rich DNA from short-read sequencing technology to the reference genome and estimating the true number of repeats in the sample genome. Fortunately, Matt Danzi on my team befriended Egor Dolzhenko at Illumina who had developed a tool called ExpansionHunter Denovo (EHDn) to do just that. Egor generously allowed us to use it before it was made publicly available. We used EHDn to detect TRs on a genome-wide scale, focusing specifically on TRs larger than ~175bp. We selected this approach because we noted a gap in the availability of information for long repeats in control genomes, while almost all non-coding repeat expansion diseases are caused by TRs of at least 175bp. Another advantage of EHDn over other available tools is its unbiased nature of detecting repetitive motifs in the genome, which means that it does not need guidance from a reference TR set and can detect motifs that differ from the reference genome.
When we got our hands on PacBio long-read sequencing data for 5 of our samples, we finally had the chance to truly test how accurate EHDn was at identifying expansions. We were delighted to see that our false positive detection rate was less than 4%. This gives us the confidence that our data is extremely reliable. We are excited to share this dataset with the greater scientific community so that we can spur a collective effort to identify novel pathogenic repeat expansions and begin to close the gap in missing heritability.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in